
Researchers have developed a groundbreaking data-driven model to predict the dehydrogenation barriers of magnesium hydride (MgH2), a promising material for solid-state hydrogen storage. This advancement holds significant potential for enhancing hydrogen storage technologies, a crucial component in the transition to sustainable energy solutions.
Hydrogen, recognized for its versatility and clean energy potential, can be produced from various renewable sources. Solid-state hydrogen storage materials, particularly MgH2, are considered prime candidates for efficient hydrogen storage due to their high storage capacity and resource abundance. However, despite extensive research over the past five decades, the material properties of MgH2 have yet to meet the performance targets set by the US Department of Energy (US-DOE).
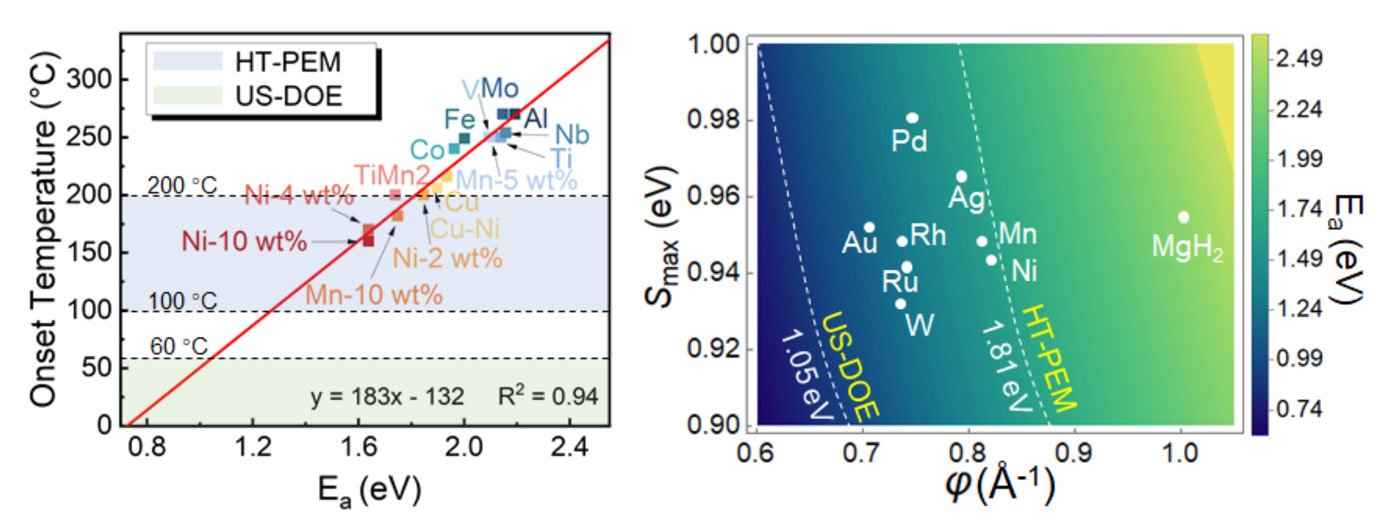
The key challenge lies in understanding the fundamental principles of solid-state hydrogen storage reactions. Current methods to assess the efficiency of hydrogen storage materials rely on dehydrogenation enthalpy and energy barriers, with the latter being particularly complex and computationally intensive to calculate. Traditional transition state search techniques, though refined over time, remain costly and time-consuming, limiting the pace of discovery and optimization.
To address this, the research team has introduced a model that predicts the dehydrogenation barriers using easily computable parameters: the crystal Hamilton population orbital of the Mg-H bond and the distance between atomic hydrogen atoms. By deriving a distance-energy ratio, the model captures the essential chemistry of the reaction kinetics with significantly lower computational demands than conventional methods.
"Our model offers a faster, more efficient way to predict the dehydrogenation performance of hydrogen storage materials," said Hao Li, associate professor at Tohoku University's Advanced Institute for Materials Research (WPI-AIMR) and corresponding author of the paper. "This allows us to bridge the knowledge gap left by experimental techniques and accelerate the development of high-performance hydrogen storage solutions."
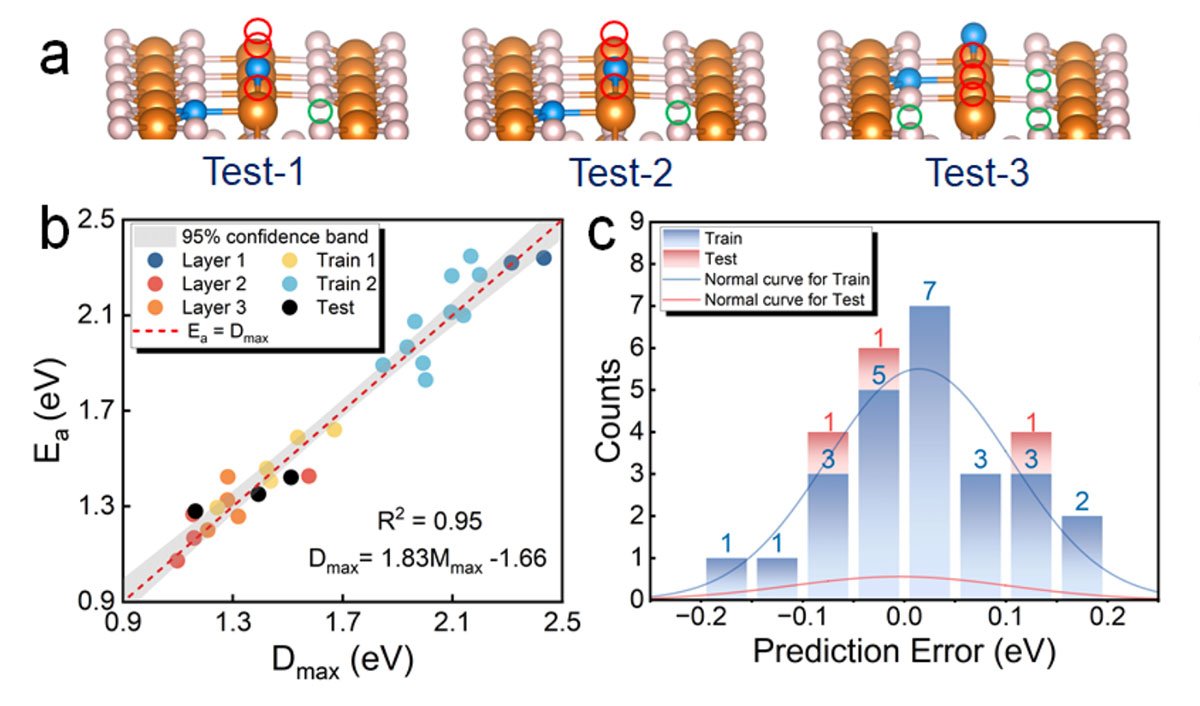
The model's predictive power was validated against typical experimental measurements, showing excellent agreement and providing clear design guidelines to enhance the performance of MgH2. This breakthrough not only propels magnesium hydride closer to the US-DOE targets but also sets the stage for broader applications in other metal hydrides.
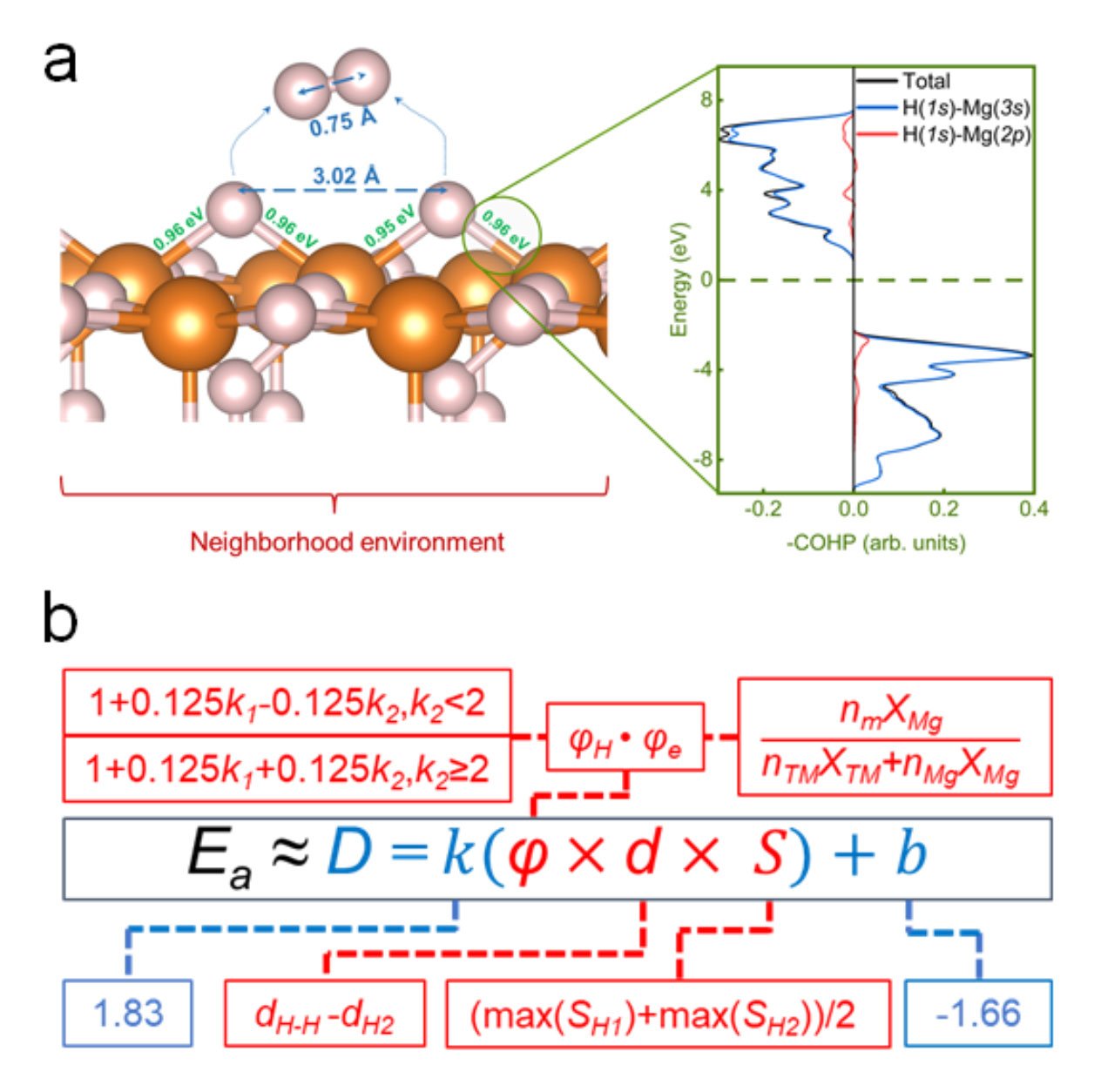
The research team plans to extend the model's application beyond magnesium-based materials. The flexibility of the model's variables allows for rapid recalibration to different metal hydrides, potentially facilitating the discovery of new composite materials and innovative solid-state hydrogen storage solutions.
"By adapting our model to various metal hydrides, we can expedite the exploration and optimization of hydrogen storage materials, paving the way for cleaner and more efficient energy systems," added Li.
- Publication Details:
Title: Picturing the Gap Between the Performance and US-DOE's Hydrogen Storage Target: A Data-Driven Model for MgH2 Dehydrogenation
Authors: Chaoqun Li, Weijie Yang, Hao Liu, Xinyuan Liu, Xiujing Xing, Zhengyang Gao, Shuai Dong, and Hao Li
Journal: Angewandte Chemie International Edition
DOI: 10.1002/anie.202320151
Contact:
Hao Li,
Advanced Institute for Materials Research (WPI-AIMR)
Email: li.hao.b8 tohoku.ac.jp
tohoku.ac.jp
Website: https://www.li-lab-cat-design.com/